Introduction
European Union (EU) Clinical Trials Regulation No 536/2014 (‘the Regulation’) has been operational since 31 January 2022. It covers interventional clinical trials of investigational medicinal products for human use and has important impacts on all aspects of trial setup, operation, and reporting in the EU and European Economic Area (EEA).
The anticipated benefits of the Regulation for the European Medicines Agency (EMA), sponsors, patients, and the public are:
- Faster, more efficient trial setup.
- Higher safety standards.
- Greater insight into trial management and outcome.
See the poster ‘Overview of Transparency Requirements for EU Clinical Trials Regulation No 536/2014’ (link) for more details and links to additional resources.
This post summarises Year 1 of the Regulation from a sponsor perspective, with a focus on transparency aspects.

Purpose of Year 1
There is a 3-year transition period for the Regulation. During Year 1 of the transition period (31 January 2022 to 30 January 2023), new EU trials could follow the earlier EU Clinical Trials Directive (CTD) or the Regulation, with the following recommendations:
• New trials expected to be ongoing after 30 January 2025 were encouraged to follow the Regulation.
• Trials ongoing under EU CTD had the option to transition to the Regulation.
• Voluntary Harmonisation Process trials had to follow the Regulation or file national submissions for substantial amendments under EU CTD.
Prior to compulsory use of the Regulation on 31 January 2023, key objectives of the Year 1 transition year were to allow all sponsors and EU/EEA Member States (MSs) to learn and adapt to the Regulation and to detect problems with new processes and the new portal (Clinical Trial Information System, (CTIS)) so that issues could be resolved before the end of the transition year.
It was recommended by EMA that sponsors at a minimum select 2 or 3 current trials to transition to the Regulation during Year 1 for experience working with the new system.
Learnings From Year 1
Metrics
Since May 2002, the EMA has published a monthly report on key performance indicators related to implementation of the Regulation (link). Information from the most recent (31 January 2023) report is summarised below.
Overall, 681 clinical trial applications were submitted in CTIS since the launch of the system on 31 January 2022 through 31 December 2022, of which 552 were initial clinical trial applications (including both new trials and trials transitioning from EU CTD), 110 were substantial modification applications, and 19 were applications for the addition of a new MS.
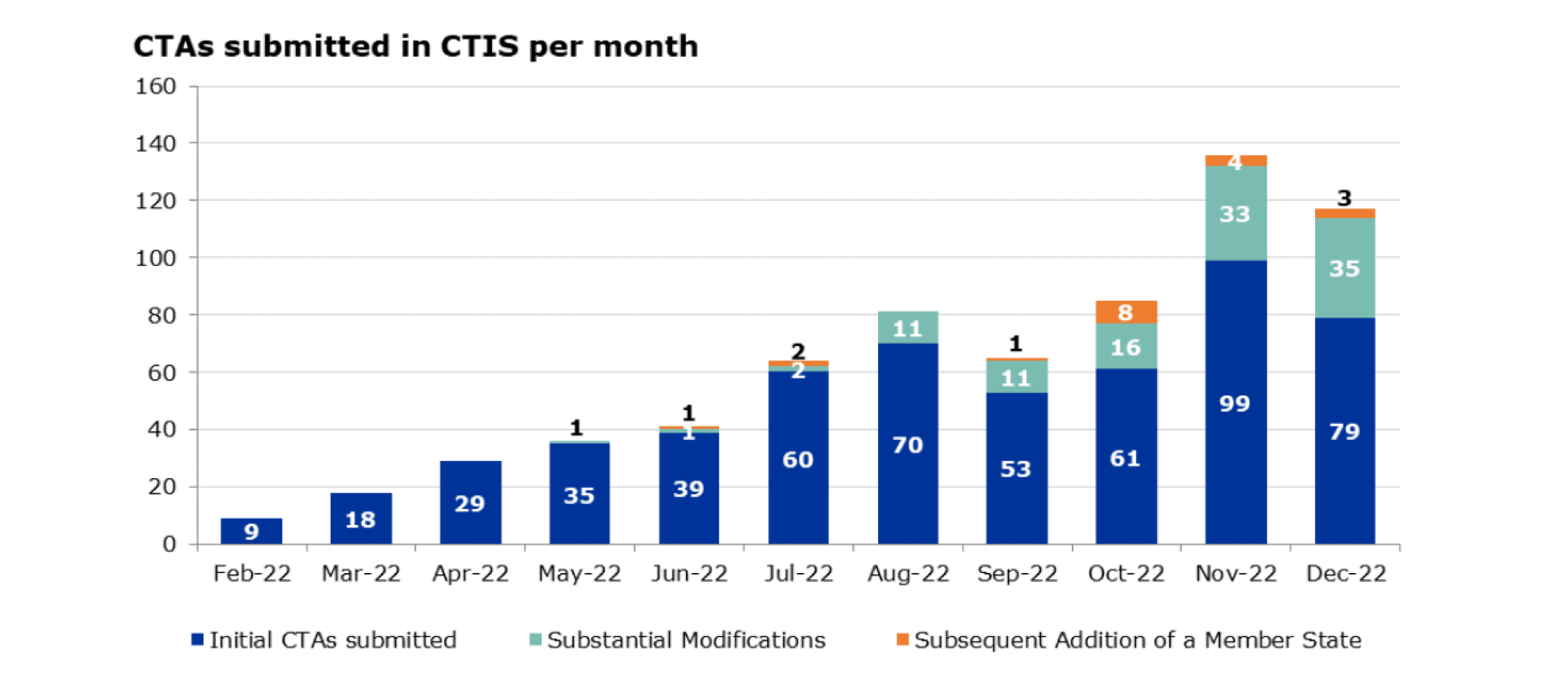
Source: European Medicines Agency, ‘Key performance indicators (KPIs) to monitor the European clinical trials environment’ EMA/2/2023, 31 January 2023 (link)
Of the 552 initial clinical trial applications through 31 December 2022, 196 have been authorised, 56 withdrawn, and 36 lapsed, with most of the rest still under evaluation.
A total of 214 of these 552 trials had a decision in CTIS of authorised, not authorised, or ended. Of these, 123 were sponsored by commercial (including pharmaceutical company) sponsors and 91 by non-commercial (including academic) sponsors. On average it took 84 calendar days to issue a decision for the 214 trials.
Guidance
On 7 April 2022, EMA published a draft “Guidance document on how to approach the protection of personal data and commercially confidential information in documents uploaded and published in the Clinical Trials Information System (CTIS)”. The EMA is in the process of preparing a final version of this guidance based on feedback received during the open consultation period that ended on 8 September 2022. The final guidance, which is expected in early 2023, will be essential to sponsors as they solidify long-term processes and strategies for ensuring proper protection of personal data and confidential commercial information (CCI) in their submission documents.
In advance of the expected final EMA guidance, 2 letters were published that are relevant to transparency under EU CTR:
• European Medicines Agency, ‘Q&A on the protection of commercially confidential information and personal data while using CTIS’ EMA/898965/2022, 12 January 2023
• Clinical Trials Coordination and Advisory Group, ‘Clinical Trials Regulation (EU) No 536/2014 in practice’ Version 01, 30 January 2023
The Agency has also published a “Clinical Trial Information System (CTIS) – Sponsor Handbook” that waslast updated in December 2022 (link).
Deferral Process and Redaction of Confidential Commercial Information
Deferral vs Redaction
To protect CCI, sponsors have the option under the Regulation to request deferral of publication of key trial information and documents up to 1, 5, or 7 years after the end of the trial in the EU/EEA depending on the type(category) of trial (see poster at link).
Tip: Sponsors should develop procedures and document templates to minimize or eliminate CCI during authoring so as to reduce reliance on deferral and redaction.
While deferral is the EMA’s preferred method for sponsors to protect CCI per current disclosure guidance (link), redaction of CCI was also allowed based on information in the EU CTR Questions and Answers (Q&A) document. Redaction was encouraged as an alternative to requesting long deferrals of publication so as to enable the earliest possible publication of trial documents. In the draft guidance on protection of CCI mentioned above, it is stated that ‘sponsors could avoid the use of the deferral mechanism in case the documents can be published at the time of decision with a minimal level of redaction needed’ (bolded word in original).
However, the Q&A letter published on 12 January 2023 (link) stated that simultaneously redacting CCI and requesting deferral for the same document or set of documents equates to over-redaction and would not be acceptable. At the same time it acknowledged that, in limited situations, some pieces of information (eg, that related to Quality) may be considered CCI even after the deferral period ends and consequently would be redacted even in documents subject to deferral requests.
Further clarity on this issue may be forthcoming in the final EMA guidance. Nevertheless, indications are that deferral is the expected method for sponsors to protect CCI under EU CTR and use of redaction must be minimised or avoided.
Acceptability of Deferral Requests
The EMA has not published detailed metrics on the number of EU CTR trial applications that included a request for publication deferral, or on the number of deferral requests that were granted by MSs.
Up to 9 August 2022, it was possible to view information—including whether deferral was requested—on the CTIS public portal for clinical trials that had a decision up to that time. However, clinical trials with any type of deferral with a decision issued after mid-August have not been published on the public portal because of the issue described further below.
Nevertheless, informal viewing of the content published prior to August 2022 and anecdotal reports from sponsors indicate that the majority if not all of deferral requests have been granted to date. Furthermore, there is no indication so far that MSs are objecting to the length of requested deferral periods or requiring shorter deferrals.
The recent Q&A letter (link) stated that it is expected that MSs will comment mainly on the trial category rather than on the sponsor’s deferral request. However, the possibility of requests from MSs for more information on proposed deferrals during review of the trial application ‘should not be excluded.’
Currently available CTR guidance requires that justifications for deferral consist of 2 parts: a justification for the category (1, 2, or 3) of the trial and a justification for the publication deferral itself. Current guidance (link) indicates that the latter is required for all 3 categories of trial. In practice, CTIS requires justification of the following:
a) The category for all types of trial;
b) The publication deferral for Category 1 trials;
c) The publication deferral for Category 3 trials that include a protocol.
Tip: Sponsors should ensure the correct trial category is selected. A multiphase or adaptive study design that appears to fall into both Categories 1 and 2 must be treated as Category 2. Emergency- situation trials where informed consent is sought after the 1st trial-specific intervention are always Category 2 or 3.
Requiring justification of publication deferral only for a limited number of trials simplifies the process of preparing justifications for sponsors. However, it is unclear if this practice will continue or if future CTR guidance will be revised to align with current CTIS practice.
While further clarity on acceptability of deferral requests could be forthcoming in the final EMA guidance, it is more likely that sponsors will learn the best way to manage deferrals through practice and experience as they move through Year 2 of the transition and beyond.
Inadvertent Publication of CCI
In July 2022 the EMA became aware that information related to some clinical trials had been prematurely published on the CTIS public portal. For clinical trials with agreed deferrals, requests for information (RFIs) from EU/EEA MSs, responses provided by sponsors, and assessment reports related to Part I and Part II were made available on the public portal, in contravention of endorsed transparency rules.
From 9 August 2022 to the end of that month, while investigation of the root cause of the issue and its resolution were ongoing, the CTIS search function allowing users to search and access published clinical trial data was deactivated. During the shutdown, verification was conducted by EMA of all trials with deferrals to confirm that only the correct information was accessible via the public domain. Previously affected trials had the correct disclosure rules applied to prevent publication of RFI, RFI responses, and assessment reports.
As an additional mitigation measure, clinical trials with any type of deferral with a decision issued after mid-August have not been published on the CTIS public portal. For new clinical trial applications in CTIS, sponsors could still request deferrals when completing the clinical trial application form and EU/EEA MSs could apply deferrals as applicable. All these clinical trials will be published on the CTIS public portal once full functionality is restored.
As of this writing the moratorium is still in effect. About 90 trials have been published on CTIS through 31 December 2022, compared to the 214 trials reported by the EMA as having a decision by that date.
Presumably as an additional measure to protect CCI, particularly in RFIs, EMA will allow sponsors to mark text with a red box outline in the “not-for-publication” version of documents to indicate what they consider to be CCI.
New Information in EU CTR Q&A Document
With the aim of informing on technical aspects of EU CTR with a view to facilitating its implementation, the European Commission has been publishing a Q&A document with regular updates. The document (currently Version 6.3, published in December 2022) covers the scope of the Regulation, different types of application, safety reporting, authorisation of investigational medicinal products, and other aspects in a dozen separate sections, several annexes, and over 160 questions. The views expressed in the document are not legally binding.
Tip: Sponsors should continue to monitor updates to the Q&A document during the transition period because it has frequently been used to clarify issues around the Regulation and to publicise possible future adjustments to guidance.
Aspects related to disclosure and transparency are mentioned across sections. Some answers that potentially impact transparency are as follows:
Question 1.21: What are the languages requirements for documents that constitute part I of the application dossier?
Answer: The language of the application dossier or parts thereof shall be determined by the (MSs). The CTR asks the (MSs) to consider using a commonly understood language in the medical field for documentation that does not go to the subject. Member States have indicated in annex II which documents from the part I (…) can be accepted in English, and what documents are (obligatory) to be submitted in other languages as well. It should be noted that translated documents adhere to the same publication rules as the original document.
Question 1.24: How are patient facing documents expected to be submitted?
Answer: Patient facing documents are documents, other than recruitment material or subject information sheets, presented to clinical trial participants during the conduct of the clinical trial. These can be questionnaires, patient diary, patient card or patient reported outcomes. Patient facing documents that are linked to the endpoints of the clinical trial shall be provided together with the protocol in part I of the clinical trial application (…). These documents will be assessed during the part I assessment.
Question 5.8: What should be included in the protocol synopsis described in Annex I, D.24?
Answer: (…) Sponsors should consider to make the synopsis understandable to a layperson.
Question 11.15: After implementation of the CTR, will EU phase 1 trials meet the CTR transparency requirements as only very limited data will be published in the EU database?
Answer: (Article) 25(6) (of the Regulation) is a shall provision and accepts trial data submission as part of a (clinical trial application) only if the referenced trial was registered publicly (including registration in the EU Clinical Trial Register) or the results have been published in a peer-reviewed manner. The last possibility is only there for trials that started before the applicability of the CTR. If a referenced trial is not registered in an (International Clinical Trials Registry Platform) database or the results are not published, the data cannot be used to support an application in the EU under the CTR, irrespective of whether it is a phase 1 trial in adults or not.
Given that new information in the Q&A document is limited in detail and it can be unclear if it constitutes a recommendation or requirement under the Regulation, it has been challenging forsponsors to assess its impact. In addition, since it was published during Year 1 while sponsors and MSs were learning to implement the new processes, it has complicated preparation of trial documentation for new and transitioning trials. Furthermore, some sponsors received RFIs reflecting upcoming Q&As in advance of publication of a new version of the latter, which can be very challenging given RFI timelines.
Outlook for Year 2
Beginning on 31 January 2023, all new EU clinical trials must follow the Regulation. Many sponsors will have already initiated or transitioned trials following the new processes, and most EU/EEA MSs will have assessed CTIS trial applications for sites in their country.
The EMA previously committed to identifying and correcting any problems with CTIS in advance of Year 2.
In Year 2, sponsors will be looking for the following regarding disclosure and transparency under the Regulation:
• Assurance that CCI in trial documents will be appropriately protected, particularly in RFIs, sponsor responses, and MS assessment reports;
• Confidence in the deferral assessment process, including whether sponsors will have an opportunity to provide further justification during the RFI process if necessary;
• Clarity on how and if redaction of potential CCI will be assessed by MSs, including in combination with deferral of publication, and whether sponsors be given an opportunity to adjust the amount of redaction in “for-publication” documents; • Clarity on how different MSs will ensure consistency in CCI protection over time across trials for a given product;
• Clarity on new transparency issues raised in the Q&A document, including…
-
Trial document language requirements across countries and implications for the cross-EU/EEA harmonisation that is one of the aims of the Regulation;
-
Need for new types of trial document not covered in current EU CTR disclosure guidance, including patient-facing documents and a lay version of the protocol synopsis;
-
How to refer to data from unregistered, unpublished Phase 1 trials in, eg, the Investigator’s Brochure;
• Clarity on whether and how to register 3rd-country trials that are part of an EU PediatricInvestigation Plan in CTIS as well as EudraCT (eg, see link);
• More information on the results disclosure process in CTIS and alignment with EMA publication policies such as Clinical Data Publication;
• Clarity on whether documentation on auxiliary medicinal products is subject to publication via CTIS.