– Written by Bhavin Busa
I’ve been meaning to share a write-up for the broader PHUSE Community. I’m a little late getting to this, but better late than never. This is an important and impactful topic for our industry, and we didn’t want to limit the insights to those who attended the conference. I have also included links to download the slides.
At the PHUSE US Connect 2025, I had the opportunity to moderate a panel featuring representatives from the European Medicines Agency (EMA), Genentech, PHUSE and others, focused on the evolving future of data submissions in the EU.
The discussion centred around the EMA’s efforts to assess whether clinical study data, in standardised formats such as SDTM and ADaM, can help speed up and improve the medicine evaluation process. Notably, while EU regulators can already request clinical study data from applicants/marketing authorisation holders (MAHs), systematic submission of such data may soon become the reality in the centralised procedure with the European Commission’s proposal for a reform of the EU pharmaceutical legislation.
Unlike other major regulatory agencies such as the FDA, PMDA or NMPA, which already require the submission of clinical study data for review, the EMA has not traditionally mandated this level of data granularity.
For a broader understanding of global regulatory requirements for data submission, the PHUSE Regulatory Environment education cluster provides a helpful overview.
A New Vision for Data Submission in Europe
Launched in 2022, the EMA Committee for Medicinal Products for Human Use (CHMP) clinical study data pilot set out to evaluate the benefits of early access to clinical datasets during the regulatory review process. The pilot’s interim report, published in late 2024 , highlighted how access to standardised datasets improved assessors’ ability to understand results, ask fewer clarifying questions and better target GCP inspections.
Eftychia Eirini Psarelli (EMA): “The pilot’s participants reported that access to clinical study data supports more confident, timely decisions – with no delay to the review timeline.”
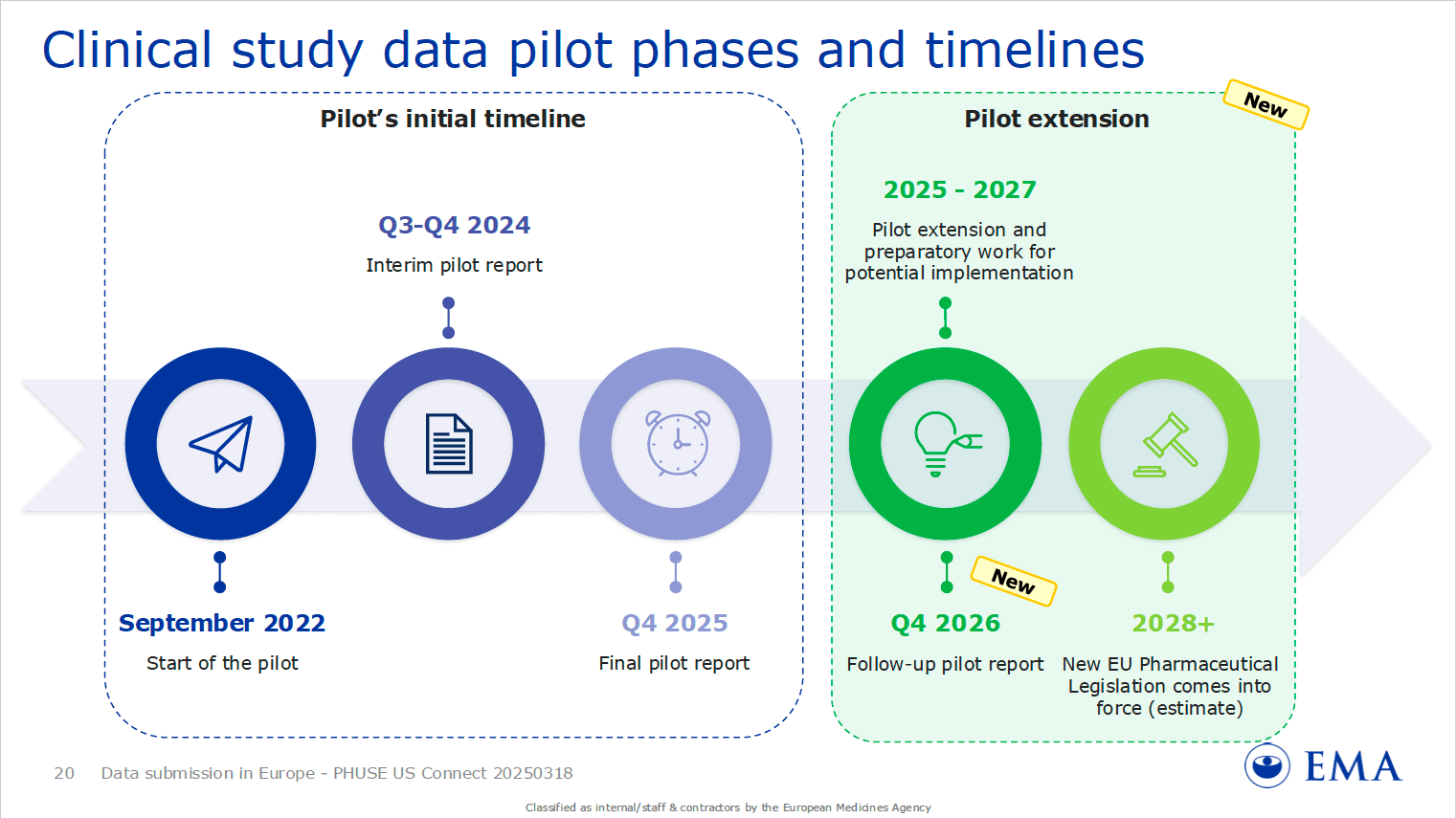 Figure 1: Visual from the slides presented by Eftychia Eirini Psarelli (EMA) detailing the EMA’s pilot phases and timelines
Figure 1: Visual from the slides presented by Eftychia Eirini Psarelli (EMA) detailing the EMA’s pilot phases and timelines
Genentech’s Firsthand Experience
Genentech shared their experience participating in the EMA CHMP clinical study data pilot, where they submitted an end-to-end R-based package originally developed for the FDA. The submission included all CDISC-compliant datasets, metadata, R code and validation documentation. EMA reviewers confirmed alignment with sponsor results and raised no additional questions during assessment.
Jingyuan Chen (Genentech): “The pilot didn’t add extra work for the data science team – it simplified the process. Our FDA-ready package was fully accepted, and communication with the EMA was smooth and constructive .” Jingyuan further mentioned: “We believe this pilot reduced the number of questions during the review procedure, with potential positive impact on the timelines.”
 Figure 2: Visual from the slides presented by Jingyuan Chen (Genentech) explaining what they prepared for FDA vs EMA requirements
Figure 2: Visual from the slides presented by Jingyuan Chen (Genentech) explaining what they prepared for FDA vs EMA requirements
PHUSE’s Role in Driving Collaboration
Sascha Ahrweiler (PHUSE Board of Director: Communications and Brand Strategy), representing PHUSE, explained how the organisation participated in the EMA’s Industry Group focusing on clinical study data to present best practices for structured data submission. PHUSE was invited by the European Federation of Statisticians in the Pharmaceutical Industry (EFSPI) to join the Industry Focus Group. At the beginning of the clinical study data pilot, PHUSE did not have EMA stakeholder status, which has since changed.
This collaboration gave PHUSE members a platform to contribute to shaping tools and frameworks that support clinical study data review by regulators. Sascha noted the EMA’s transparency and eagerness to learn from the FDA’s experience while forging a distinct European model . He expressed sincere appreciation for the opportunity and acknowledged that this was an exemplary collaborative initiative spearheaded by the EMA.
Sascha Ahrweiler (PHUSE): “I’m grateful for this true collaborative approach the EMA chose to get the industry involved. We’re not just reacting – we’re building the future of regulatory submissions together.”
 Figure 3: Visual from the slides presented by Sascha Ahrweiler (PHUSE)
Figure 3: Visual from the slides presented by Sascha Ahrweiler (PHUSE)
What’s Next: Visualisation, Automation and Open-Source Tools
Looking ahead, the EMA’s efforts are focused on:
- Developing open-source tools (e.g. R Shiny) for dynamic analysis and visualisation available to all the assessors within the European Medicines Regulatory Network (EMRN), including:
- Standardised, CDISC-compliant visualisations
- Dynamic filters for variables and populations
- Automating validation and metadata characterisation
- Collaborating internationally with the FDA and the PMDA to reduce burden on applicants
- Integrating these tools into the EMA’s IT infrastructure.
Marie Orre (EMA): “We envisage assessors, even those without coding expertise, gaining insights from the data through using accessible, validated interactive tools.”
 Figure 4: Visual from the slides presented by Marie Orre (EMA) discussing interactive and open-source tools for future integration with the EMA’s data storage and analysis infrastructure
Figure 4: Visual from the slides presented by Marie Orre (EMA) discussing interactive and open-source tools for future integration with the EMA’s data storage and analysis infrastructure
This PHUSE panel made it clear: the EMA CHMP clinical study data pilot is leading with openness, industry collaboration, and a strong focus on modernising the review process. The clinical study data pilot is paving the way for a more intelligent, efficient and harmonised regulatory model across Europe.
Audience Q&A Highlights
Will participation in the EMA CHMP clinical study data pilot impact on review timelines for applicants?
“No. We’ve demonstrated that assessors can conduct clinical study data analysis without introducing delays. The overall review timeline remains unchanged.” – EMA Representative
Are there any specific language requirements for clinical study data or metadata submissions?
“No. English is the only required language for centralised submissions. There are no translation or localisation requirements for metadata.” – EMA Representative
How does this initiative align with other regulators such as the FDA or the PMDA?
“We’ve engaged in meaningful dialogue with both agencies. Like the PMDA, the EMA is following a stepwise pilot approach. We’re committed to minimising divergence and supporting harmonisation where possible.” – EMA Representative
What about the use of open-source tools such as R or Python in submissions?
“R is fully accepted, and we’re open to Python and other open-source platforms as long as the tools are validated and usable within our IT infrastructure.” – EMA Representative
“Our R-based submission was well received. We included validated packages and code documentation, and EMA reviewers had no issues.” – Jingyuan Chen, Genentech
Could we eventually move towards fully machine-readable submissions and fewer PDFs?
“In the near term, we don’t expect to eliminate submission documents entirely. But we do envisage adding dynamic visualisations and interactive summaries alongside traditional components.” – EMA Representative
“This is a real opportunity to align how we present data and possibly reduce document-heavy submissions in the future.” – Sascha Ahrweiler, PHUSE
Links to Presentations
Acknowledgements
I sincerely thank my panellists – Eftychia Eirini Psarelli (EMA), Jingyuan Chen (Genentech), Marie Orre (EMA) and Sascha Ahrweiler (PHUSE) – for their thoughtful contributions during the session and for their valuable feedback on this article. A special thank you to Chris Price (PHUSE) for his support in shaping this panel and for reviewing the content.
