By Varun Debbeti, Principal Statistical Programmer, SpringWorks Therapeutics
Every Phase III trial ends in a mountain of tables, figures and listings – 150 or so at organisations that keep their reports lean; several hundred at those that don’t. A 2025 structured review on medRxiv puts the typical range at 200 to 500. Whatever the exact count, that volume hides a massive operational bottleneck. Independent double programming adds 1.6 to 2.0 times the original effort, and iterative spec revisions stretch timelines by 20 to 40 per cent. These numbers reflect standard practice, and they are an unsustainable drain on budget and time-to-market. Just as we merge overlapping clinical systems to reduce friction, we must do the same with our data standards.
CDISC has spent the last three years releasing the building blocks to fix this. USDM standardises the study definition. ARS standardises the analysis specification. CORE turns CDISC conformance rules into executable code. Each one is solid on its own. The wiring between them is what no one has built yet fully. This post makes the case for treating these three as one connected pipeline rather than three parallel projects, and for statistical programmers to push their organisations in that direction.
Three standards, three different problems
USDM v3.0 was released in April 2024, and v4.0 followed in 2025. It replaces the Word protocol with a structured, machine-readable description of arms, schedules, endpoints and inclusion criteria. ARS v1.0 was published in April 2024 and does the equivalent for analyses: every table’s structure, population, statistical method and layout captured in metadata. CORE turns the CDISC conformance rule library into executable code that any party can run with identical results. These three standards, when read separately, look useful but incremental. The value is in the handoff between them.
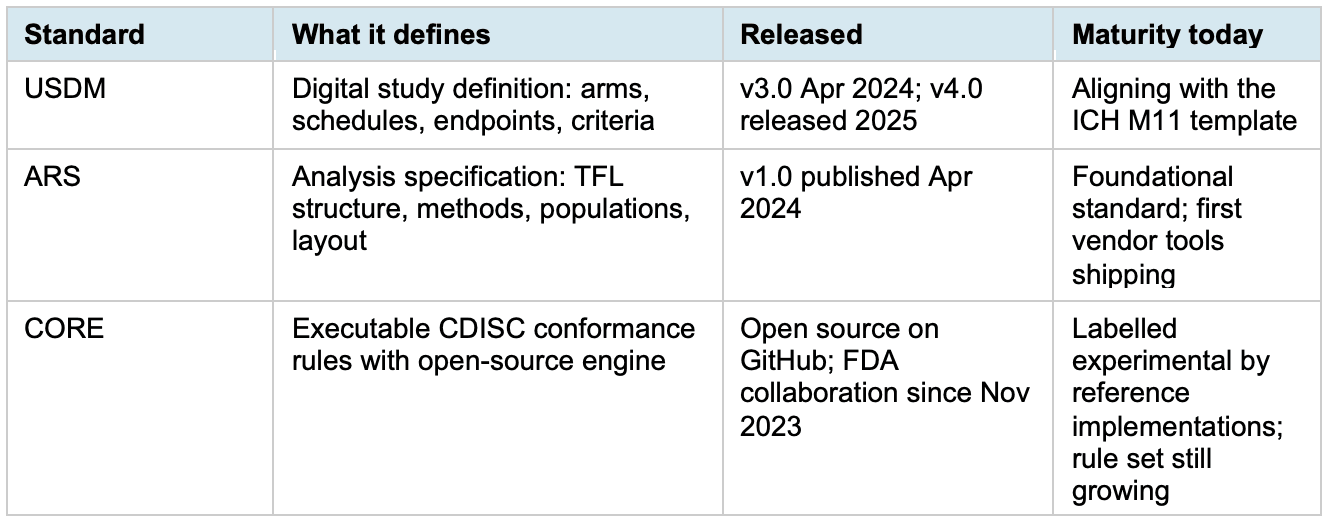
Table 1. USDM, ARS and CORE at a glance.
CDISC is already building the pipeline
This is no longer a thought experiment. In March 2025, CDISC launched 360i, an initiative whose explicit goal is standards-driven automation across the data life cycle, from study design through analysis to submission. Phase 1 linked digital protocols to biomedical concepts to auto-generate eCRFs, SDTM specs and Define-XML. Phase 2, underway in 2026, extends the chain into analysis, exactly where ARS lives. 360i is the institutional proof that these standards are meant to interlock, not sit in separate working groups.
Picture a study where the USDM file is the source of truth, driving EDC setup, the ADaM specifications and the TFL shells. The TFL shells themselves are ARS metadata, so the analysis dataset specs and the output programs read from one file. CORE rules run against SDTM and ADaM at every milestone, not only at submission, with results visible within minutes.
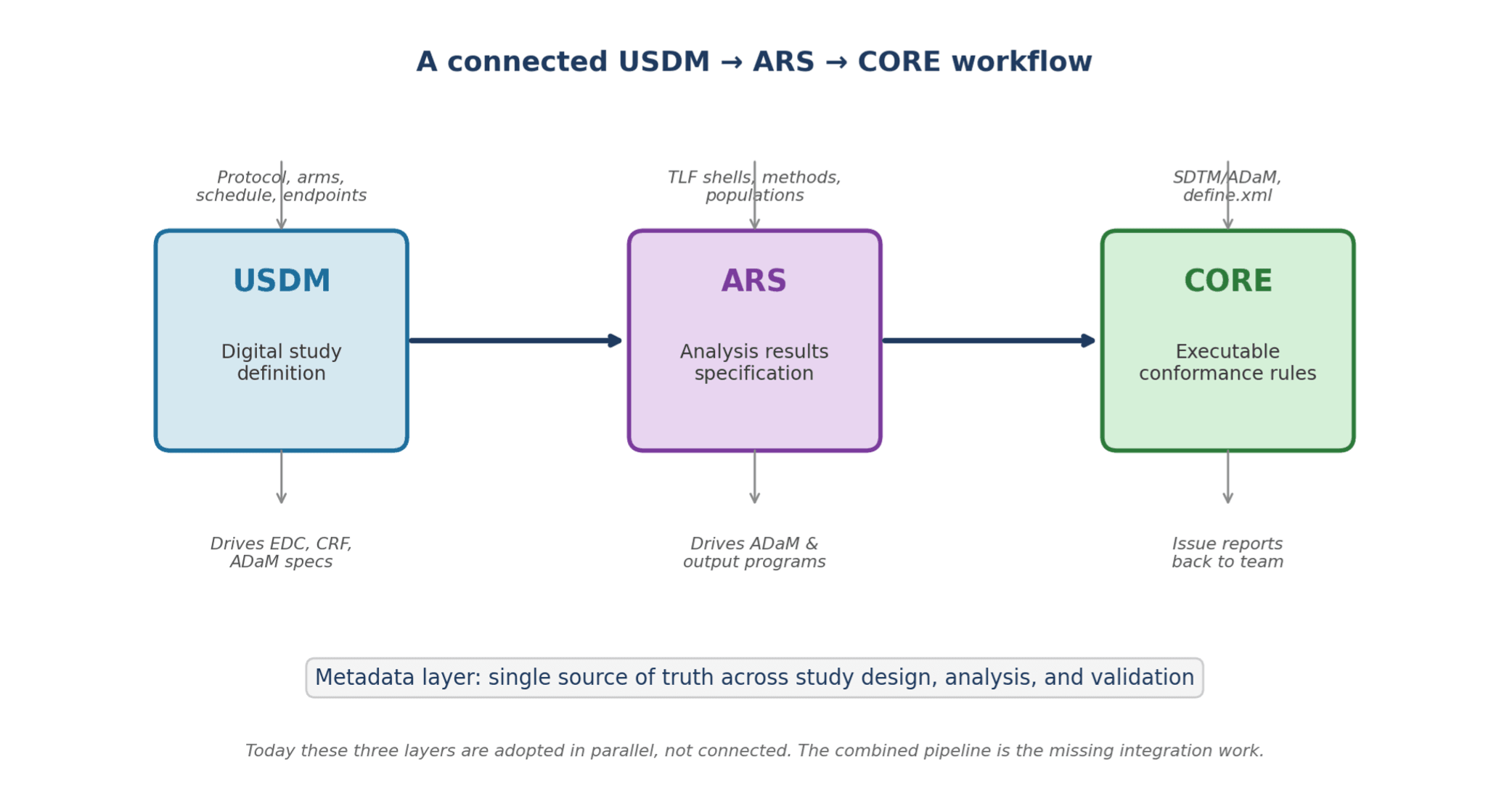
Figure 1. The connected pipeline. Each standard targets a different layer of the same study workflow.
A worked example
To test whether this holds up in practice, I built an open-source ARS-CORE Workbench at clinstandards.org. It is a six-step browser pipeline. A programmer designs TFL shells using ARS concepts (AnalysisSet, GroupingFactor, Method, Output), including oncology efficacy templates for ORR, PFS, DOR and OS. Those shells link to an ADaM spec, where a cross-spec check confirms every variable where a shell reference actually exists in the datasets, catching breaks before any code runs. The merged bundle is handed to an AI to generate SAS or R, and CDISC CORE rules validate the datasets before everything is packaged into an audit trail. The value is not just the AI. It is that the structured, deterministic nature of ARS metadata, rather than free-form prose, gives the model an unambiguous specification to work from, which yields far more accurate and reproducible results end to end.
What programmers actually gain
Return to that output count, whether it runs to a lean 150 or several hundred. If even 60 per cent of those tables can be built from ARS specs reused across studies in a therapeutic area, the second study no longer requires rebuilding the same shells. A standard AE summary or an oncology OS table, once specified, becomes a reusable asset. The double-programming overhead drops because the metadata, not free-form code, is what gets validated. CORE running continuously means the find-fix loop on conformance issues collapses from weeks to hours.
For a programmer, more of the work moves upstream into metadata authoring and review: less time on production SAS, more on the design of the analysis itself.
Current limitations
Three caveats are worth naming. CORE is still labelled experimental by reference implementations such as Pinnacle 21 Community. The rule set is limited, false negatives are expected, and the documentation tells you to use a production-grade validator for actual submissions. The CDISC–FDA collaboration to incorporate FDA Business Rules into CORE began in November 2023 on a three-year term, so a production-ready version is still ahead of us.
USDM adoption depends on whether the protocol authoring side moves. Most sponsors still produce protocols as Word documents. ICH M11 will likely be the forcing function, since its template aligns with USDM, but companies waiting for the mandate will be late to the integration work that follows.
ARS reuse is real but uneven. A safety summary generalises well; a bespoke biomarker analysis resists standardisation. The first studies in a program pay an authoring tax that later studies recover.
What to do this year
PHUSE Working Groups cover each standard in depth. What we lack are integration patterns: reference architectures, open templates and worked examples that show one USDM study definition flowing through ARS-specified analyses to CORE-validated outputs. 360i is producing some of these; the community should pressure-test them.
Sponsors and CROs invested in one standard should ask what it takes to connect to the next. Programmers should learn the metadata layer of all three, not the syntax of any one. Vendors should publish how their tools consume USDM and ARS, not only SDTM and ADaM.
The 1.6 to 2.0 validation multiplier and the 20 to 40 percent timeline penalty will not disappear from any single tool or standard. They disappear only when organisations design these three standards to operate as one integrated pipeline.
References
- Yan, J., Zhang, J., & Tian, T. (2025). Automation in Clinical Trial Statistical Programming: A Structured Review of TLF Generation, Validation Frameworks, and AI/ML Integration (2020–2025) | medRxiv
- CDISC. CDISC 360i: The Art of the Possible. cdisc.org/standards/cdisc-360i
- CDISC. Unified Study Definitions Model (USDM). cdisc.org/ddf
- CDISC. Analysis Results Standard. cdisc.org/standards/foundational/analysis-results-standard
- CDISC. Research Collaboration to Incorporate FDA Business Rules into CORE. Press release, 16 January 2024. cdisc.org/core
- Pinnacle 21 Community documentation on CDISC Open Rules Engine support (Certara). pinnacle21.com
About the author
Varun Debbeti is a statistical programming leader with 13 years of experience across the pharmaceutical, biotechnology and CRO sectors, specialising in NDA, BLA and EMA regulatory submissions. Varun is the founder and lead developer of clinstandards.org, a resource hub for clinical data standards, AI tools, and education for the programming community, where he maintains an open-source ARS-CORE Workbench exploring combined USDM, ARS and CORE workflows.